Infant with ultra-rare genetic disease receives unprecedented personalized treatment
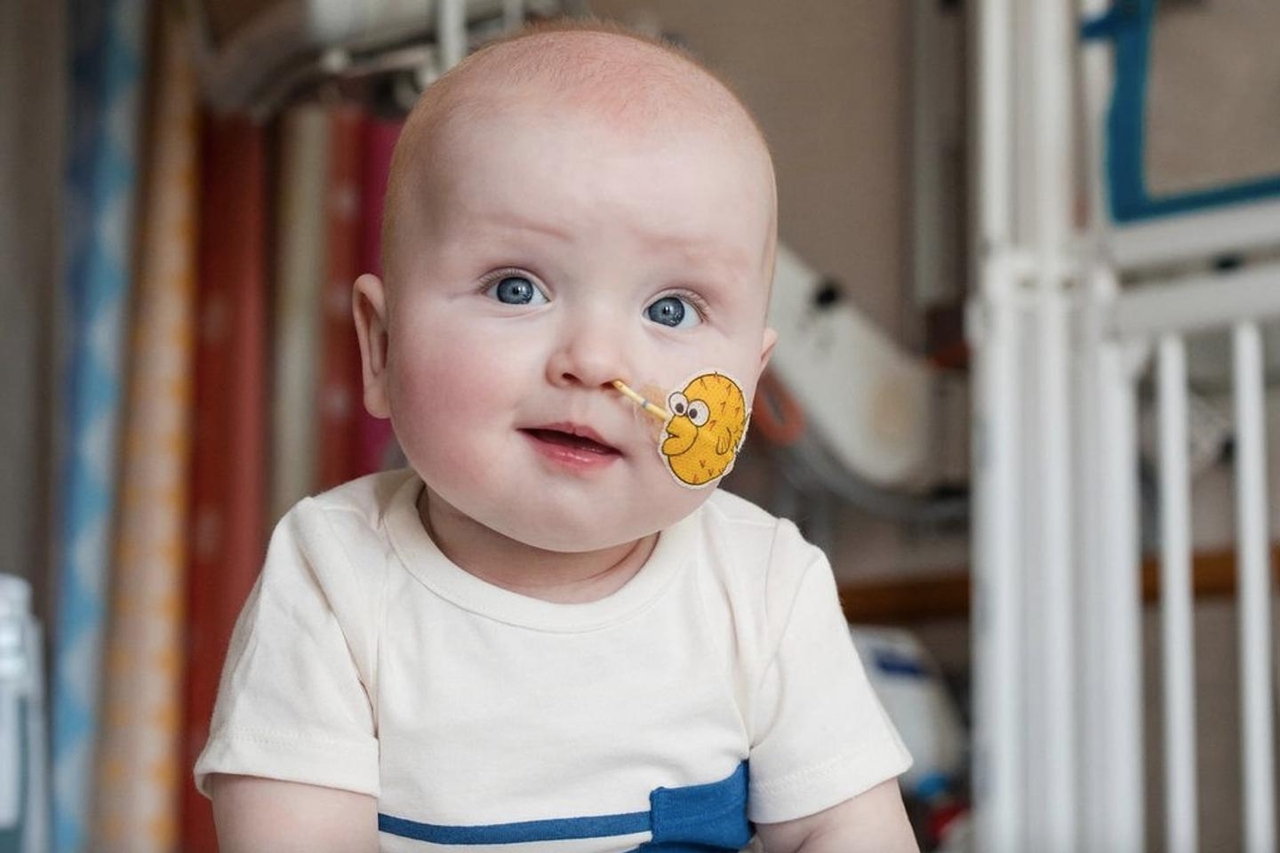
This is the story of a little boy suffering from a severe and extremely rare genetic disease , who benefited from a personalized genome editing treatment at the age of approximately 7 and 8 months. It consisted of modifying a single letter (base) in the DNA to correct the anomaly responsible for this pathology.
The particularly promising preliminary results of this innovative treatment are reported in an article published on May 15, 2025, in the American medical weekly The New England Journal of Medicine (NEJM) . This publication represents a major step in the evolution of personalized therapies for rare and ultra-rare congenital metabolic anomalies.
The child has the neonatal form of a disease called carbamoyl phosphate synthetase 1 (CPS1) deficiency. This rare condition is one of the inborn errors of metabolism, characterized by an enzyme malfunction that disrupts cellular metabolism.
Fatal disease in 50% of cases in the first months of lifeThe incidence of CPS1 deficiency is estimated at 1 case per 800,000 to 1.3 million births. This disease is fatal in approximately half of infants during the first months of life. It is caused by mutations in the gene that encodes CPS1, an enzyme found in liver cells. More specifically, in hepatocytes, it is located in mitochondria, the organelles that provide the energy needed for cellular activity.
CPS1 deficiency disrupts the urea cycle, a series of biochemical reactions that converts ammonia, a product of protein breakdown, into urea, which is excreted in the urine.
This disease typically manifests as a very high level of ammonia in the blood. Massive hyperammonemia most often appears within 24 to 48 hours of birth. It is toxic to the brain and can lead to coma or even death without prompt treatment.
Treatment options are limited: dialysis to reduce plasma ammonia, administration of a nitrogen-scavenging drug, a low-protein diet, and, later, a liver transplant. However, the neurological prognosis often remains poor.
Basic Editing: A DNA-Free CorrectionRapid analysis of the child's genome revealed a paternally inherited point mutation in the CPS1 gene, corresponding to a single erroneous letter. To correct it, the team of Kiran Musunuru and Rebecca Ahrens-Nicklas at the Children's Hospital of Philadelphia used a strategy called base editing . This work involved the collaboration of several academic and industrial partners, including the Innovative Genomics Institute at the University of California, Berkeley, Massachusetts General Hospital–Harvard Medical School (Boston), Aldevron (Fargo, North Dakota), and Acuitas Therapeutics (Vancouver, Canada).
This technique allows the modification of a single letter (or base) in the DNA of a living cell, without cutting the double helix. Highly targeted and precise, it corrects the mutation responsible for interrupting protein synthesis. Once the genetic sequence is restored, the cell can once again produce a complete, functional protein.
Quick reminder: the DNA alphabet is based on four bases: A (adenine), T (thymine), C (cytosine) and G (guanine), which pair up in pairs: A with T, C with G. Each pair constitutes a "rung" of the helical ladder formed by the DNA molecule.
Rewrite a single letter in DNA in a precise and targeted mannerThe researchers used an adenine base editor, a tool capable of acting directly at the site of the mutation to correct the incorrect letter, without cutting the DNA. Specifically, this editor causes an A–T (adenine-thymine) base pair to eventually become a G–C (guanine-cytosine) pair.
Base editing is a more precise technique than traditional CRISPR–Cas9 editing, which acts a bit like molecular scissors, cutting the DNA before the cell repairs the break. These cuts can sometimes cause unforeseen side effects, called “off-target effects.” In contrast, base editing works like a “precision genetic eraser”: it changes a single letter of the genome without ever cutting the DNA double helix. In other words, it rewrites a single, precisely targeted letter to correct the base pair carrying the disease-causing mutation.
Finding the exact location to correct using a “guide RNA”This infant carried two mutations in the CPS1 gene, one from his father's side and one from his mother's side. The researchers decided to correct the pathogenic mutation inherited from his father (a single A had been substituted with a G).
This base-editing system includes a CRISPR-derived enzyme called Cas9 fused to an adenosine deaminase, which can chemically modify a defective letter in DNA. To ensure this tool acts in the right place, it is guided to the mutation by a second component: a guide RNA, a small fragment of RNA used to precisely target the mutation.
The guide RNA directs Cas9 to this target sequence, which is located in close proximity to a short recognition motif, called a PAM, which is essential for Cas9 to bind. Once firmly attached to the DNA, the tool triggers the chemical conversion of the incorrect base into the correct one. Adenosine deaminase then converts the letter A (adenine) into inosine, which is then interpreted as a G (guanine) by the cellular machinery. This process thus converts an A–T base pair into a G–C pair.
But not all guide RNAs are created equal: you need to choose one that positions the base-editing tool so that the adenosine deaminase is perfectly positioned and oriented to convert the letter A to a G, once the tool is securely attached to the target DNA sequence.
The researchers tested around thirty guide RNAs to find the one that best recognized specific DNA patterns while placing Cas9 in the best position. A truly masterful work.
Development of personalized genetic treatment in record timeSeveral base editor–guide RNA combinations were constructed and tested one by one to identify the one that corrects the mutation with the greatest efficiency and precision. But it's not simple: these tests require the use of cells carrying the mutation, and ideally cells of the same type as those affected by the disease, in this case liver cells. However, cultivating this type of cell in the laboratory is often difficult, and for some rare diseases, it can take several months, or even a whole year, before having the cells needed to carry out these experiments.
To test the different combinations of their base-editing tool, the researchers inserted the mutated version of the infant's CPS1 gene into the DNA of a human liver cell line. Creating this cell line and selecting the best adenine base editor–guide RNA combination was accomplished in just two months—an exceptional turnaround time.
Preclinical studiesBefore treating the infant, the researchers conducted preclinical studies on human cells, transgenic mice, and monkeys. This work showed that at low doses, the experimental treatment enabled effective correction of the targeted mutation (up to 42% of cells corrected in the mouse livers), without significant clinical toxicity. Safety analyses did not detect any off-target editing in the treated human hepatocytes. These results made it possible to determine an initial dose deemed safe for initial administration in children.
An application for approval for this experimental treatment was submitted to the Food and Drug Administration (FDA), which gave its approval a week later.
Anticipating a possible risk of an immune response against the entire CPS1 protein, which the patient's body had probably never produced, and therefore the production of antibodies, the doctors chose to administer preventive immunosuppressive treatment to the infant (sirolimus then tacrolimus), avoiding corticosteroids, drugs likely to raise ammonia levels in patients with CPS1 deficiency.
While the blood ammonia level had reached 1,000 micromoles/L during the first 48 hours of life, it fell back to 23 micromoles/L after the first injection of the base-editing treatment.
A first injection of the basic editing tool, then a secondKJ, the initials of the baby's first name, inspired the researchers to name the guide RNA, encapsulated in lipid nanoparticles, "kayjayguran." The adenine base editor (ABE) was named "abengcemeran." The treatment itself was dubbed "k-abe" (for short).
On February 25, 2025, the experimental k-abe treatment was administered intravenously to this infant, then approximately 7 months old, at a total dose of 0.1 mg of RNA per kilogram of body weight. After this injection, dietary protein intake could be increased, sometimes beyond the recommendations for chronological age, as the child was born prematurely (at 35 weeks of gestation).
A second infusion of k-abe (0.3 mg RNA/kg) was administered 22 days after the first, as the initial biochemical correction was deemed incomplete. Despite the occurrence of two viral infections (each with diarrhea and vomiting), the child did not experience a hyperammonemic crisis and was able to continue his protein diet without interruption. The decrease in ammonia levels, which stabilized at 13 micromoles/L, indicates an effective restoration of enzymatic activity.
Two weeks after the second infusion, the dose of glycerol phenylbutyrate, a drug used to reduce high ammonia levels in urea cycle disorders, was halved without significant adverse effects. The child gained weight and his neurological condition remained stable.
Limits and perspectivesAlthough the authors observed a stabilization of the infant's clinical condition , the main limitation of this study is the short follow-up, which does not allow for a full assessment of long-term safety and efficacy, nor for prediction of the child's neurological evolution. In addition, no direct evidence of gene correction in the liver could be obtained, as a liver biopsy, an invasive procedure, was too risky for this toddler.
Several points remain to be clarified, including the duration of the therapeutic effect, the actual proportion of modified liver cells, and the potential risks of off-target effects or immune reactions. Prolonged follow-up is therefore essential to answer these questions.
The authors of the NEJM article believe it would be crucial to develop minimally invasive methods to verify the success of the base-editing strategy, thus avoiding the risks associated with liver biopsy. These methods could include blood tests targeting specific markers or in-depth quantitative analyses of metabolites. They conclude that, although this treatment was developed in an emergency situation for a devastating neonatal metabolic disease, they anticipate that "the rapid deployment of personalized gene-editing therapies will soon become standard practice for a large number of genetic diseases."
These promising results raise hope, but to become a reliable and widely usable therapy, the technique still needs to progress, warn Andrea Gropman (St. Jude Children's Research Hospital, Memphis) and Alexis Komor (University of California, San Diego) in an accompanying editorial. According to them, "translating from proof of concept to standard treatment will require rigorous ethical and regulatory oversight. For patients with CPS1 deficiency and other similar ultrarare diseases, these results are hopeful, but still require validation through the treatment of a second patient, a third, and beyond."
One thing is certain for Peter Marks, former director of the Center for Biologics Evaluation and Research (CBER) at the FDA and signatory of a second editorial in the NEJM on the progress in the development of therapies designed and developed specifically for a single patient ( N-of-1 therapy , in English): "More than 7,000 rare diseases affect approximately 30 million people in the United States and 300 million worldwide. While not all of them are eligible for current genome editing technologies, several hundred, even thousands, could be thanks to a base editing approach similar to that used by Kiran Musunuru's team ."
During his nine years at CBER, Peter Marks was a staunch supporter of rare disease research, overseeing the approval of 22 gene therapies. His unexpected departure from the FDA last March, however, cast a shadow over the regulatory future of cell and gene therapies. Despite this uncertainty, he remains optimistic about the progress being made: "While more work is needed to reproducibly deliver gene editing tools to tissues other than the liver, advances are already underway to target bone marrow, T cells, and the brain."
Little KJ , Kyle and Nicole Muldoon 's fourth child, will be 10 months old on June 1st. His parents never imagined their critically ill child would become the world's first patient to receive a personalized base edit, an innovative approach to trying to cure his dreaded ultra-rare genetic disease.
To find out more:
Musunuru K, Grandinette SA, Wang X, et al. Patient-Specific In Vivo Gene Editing to Treat a Rare Genetic Disease . N Engl J Med. 2025 May 15. doi: 10.1056/NEJMoa2504747
Gropman AL, Komor AC. Personalized Gene Editing to Treat an Inborn Error of Metabolism . N Engl J Med. 2025 May 15. doi: 10.1056/NEJMe2505721
Marks P. Progress in the Development of N-of-1 Therapy . N Engl J Med. 2025 May 15. doi: 10.1056/NEJMe2505704
Tsuchida CA, Wasko KM, Hamilton JR, Doudna JA. Targeted nonviral delivery of genome editors in vivo . Proc Natl Acad Sci US A. 2024 Mar 12;121(11):e2307796121. doi: 10.1073/pnas.2307796121
Musunuru K, Chadwick AC, Mizoguchi T, et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates . Nature. 2021 May ;593(7859):429-434. doi:10.1038/s41586-021-03534-y
Contribute
Reuse this contentLe Monde